Un estudi identifica noves funcions del gen que causa la malaltia de Machado-Joseph
El treball, publicat a la revista Cell Reports, està liderat per la Dra. Gemma Marfany, cap del grup Genètica Molecular Humana II de l'IRSJD, catedràtica del Departament de Genètica, Microbiologia i Estadística i investigadora de l'Institut de Biomedicina de la UB (IBUB) i del Centre d'Investigació Biomèdica en Xarxa de Malalties Rares (CIBERER).
L'atàxia és una malaltia minoritària d'origen genètic caracteritzada per alteracions neuromusculars degudes a la pèrdua selectiva de neurones al cerebel, l'òrgan del nostre sistema nerviós encarregat del control del moviment i de l'equilibri. L’equip investigador ha identificat noves funcions del gen ataxina 3 (ATXN3) -causant de la malaltia de Machado-Joseph, el tipus d'atàxia més freqüent- en el desenvolupament dels fotoreceptors de la retina.
Segons les investigadores i investigadors, aquests resultats són rellevants no només per aprofundir en les causes moleculars de l'atàxia i en el disseny de potencials teràpies contra aquesta malaltia, sinó també per comprendre altres malalties, com ara les degeneracions maculars.
La malaltia de Machado-Joseph, també anomenada atàxia espinocerebel·losa de tipus 3, està causada per mutacions dominants de guany de funció del gen ATXN3. Aquestes mutacions indueixen la formació d'agregats neurotòxics que comporten la mort progressiva de les neurones del cerebel. No obstant això, se sap molt poc de quina és la funció bàsica del gen ATXN3. En aquest context, l'equip de treball investiga des de fa més de vint anys les causes genètiques de les malalties hereditàries de la retina, un teixit neurosensorial que forma part del sistema nerviós central i està encarregat de la visió.
“En treballs previs, el nostre grup ja havia descobert que ATXN3 s'expressa d'una manera rellevant a la retina del ratolí adult, però també durant el desenvolupament d'aquest teixit, així que ens vam proposar esbrinar quina era la funció d'aquest gen a la retina”, explica Gemma Marfany.
Alteracions en l'estructura de la retina
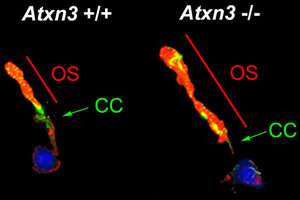
Amb aquest objectiu, els investigadors van fer diferents experiments en què van silenciar ATXN3 en embrions de peix zebra i, posteriorment, van analitzar els efectes d'inutilitzar aquest gen en un ratolí modificat genèticament. Segons els investigadors, els resultats mostren que l'eliminació d'ATXN3 causa greus alteracions en l'estructura retiniana. Així, han pogut identificar una nova funció de la proteïna d'aquest gen.
“Els organismes model analitzats, tant peixos com ratolins, presentaven una elongació del cili dels fotoreceptors, que són els orgànuls on es reben els fotons de llum i que converteixen l'energia lumínica en elèctrica”, descriu Gemma Marfany. “Qualsevol alteració en aquests cilis -continua la investigadora- altera la funció dels fotoreceptors i pot provocar-ne la mort i, per tant, causar ceguesa”.
A més, la manca de la proteïna ATXN3 afecta l'epiteli de la retina, una capa de cèl·lules pigmentades que apareixen a l'exterior de la retina i que s'encarrega de fagocitar els extrems dels cilis per permetre que es renovin cada dia. Segons el nou estudi, sense el gen ATXN3 s'alenteix aquesta fagocitosi, fet que altera aquesta renovació necessària per al funcionament de la retina.
“Totes aquestes alteracions s'expliquen si ATXN3 regula les proteïnes necessàries per a la formació i el creixement dels microtúbuls que travessen interiorment tota la cèl·lula i que fan de vies de tren per on es transporten les proteïnes requerides per a la formació del cili i per a la correcta funció de la fagocitosi”, descriu la investigadora.
Impacte en altres malalties de la visió
La importància d'aquests resultats significa un pas més en la comprensió de les causes moleculars de malalties rares com l'atàxia, però també d'altres malalties.
“L'estudi aporta noves claus sobre com es regulen i modulen funcions molt bàsiques a les neurones, entre les quals trobem els fotoreceptors. L'alteració d'aquestes funcions contribueix a moltes altres malalties de la vista, com ara les degeneracions maculars, un trastorn ocular que destrueix lentament la visió, i que afecta un percentatge alt de la població”, conclou Gemma Marfany.
En el treball també hi han participat els experts Vasileios Toulis, Sílvia García Monclús, Carlos de la Peña Ramírez, Rodrigo Arenas Galnares, Josep F. Abril i Alejandro Garanto, així com investigadors de la Universitat de Michigan (Estats Units) i de la Universitat Estatal de Wayne (Estats Units).
Font d’informació
Un estudi de la UB identifica noves funcions del gen que causa la malaltia de Machado-Joseph. Universitat de Barcelona.
La importància d’aquests resultats significa un pas més en la comprensió de les causes moleculars de malalties rares com l’atàxia, però també d’altres malalties.
